Research
Battery Materials
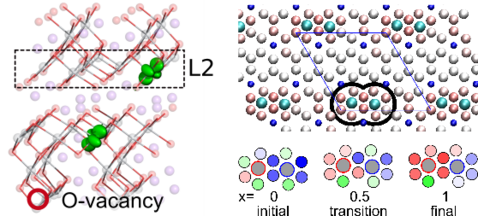
Next to the problems of the sustainable generation of energy, humanity also faces severe challenges when it comes to storing the such created energy. While electrochemical batteries have been around for over 200 years, they still lack the energy densities and long-term stabilities of e.g. fossil energy carriers. To overome these limitations a number of innovative concepts such as, for example, all solid-state batteries are being investigated all over the world. Most of this work comes down to finding the correct material and understanding the microscopic mechanisms at play in order to exploit them for maximum efficiency. By their very nature, most of these processes boil down to the movement of charge carriers, either electrons or ions. The methods our group has perfected over the years are thus perfectly suited to investigate the basic mechanisms of operation of a battery material.
This microscopic insight can then be used to improve already known materials, e.g. through the targeted engineering of defects in certain patterns to increase the mobility of charge carriers. This not only can lead to improved conductivities of materials but also to increased stabilities by avoiding locally charged trap states.
Computational Materials Design
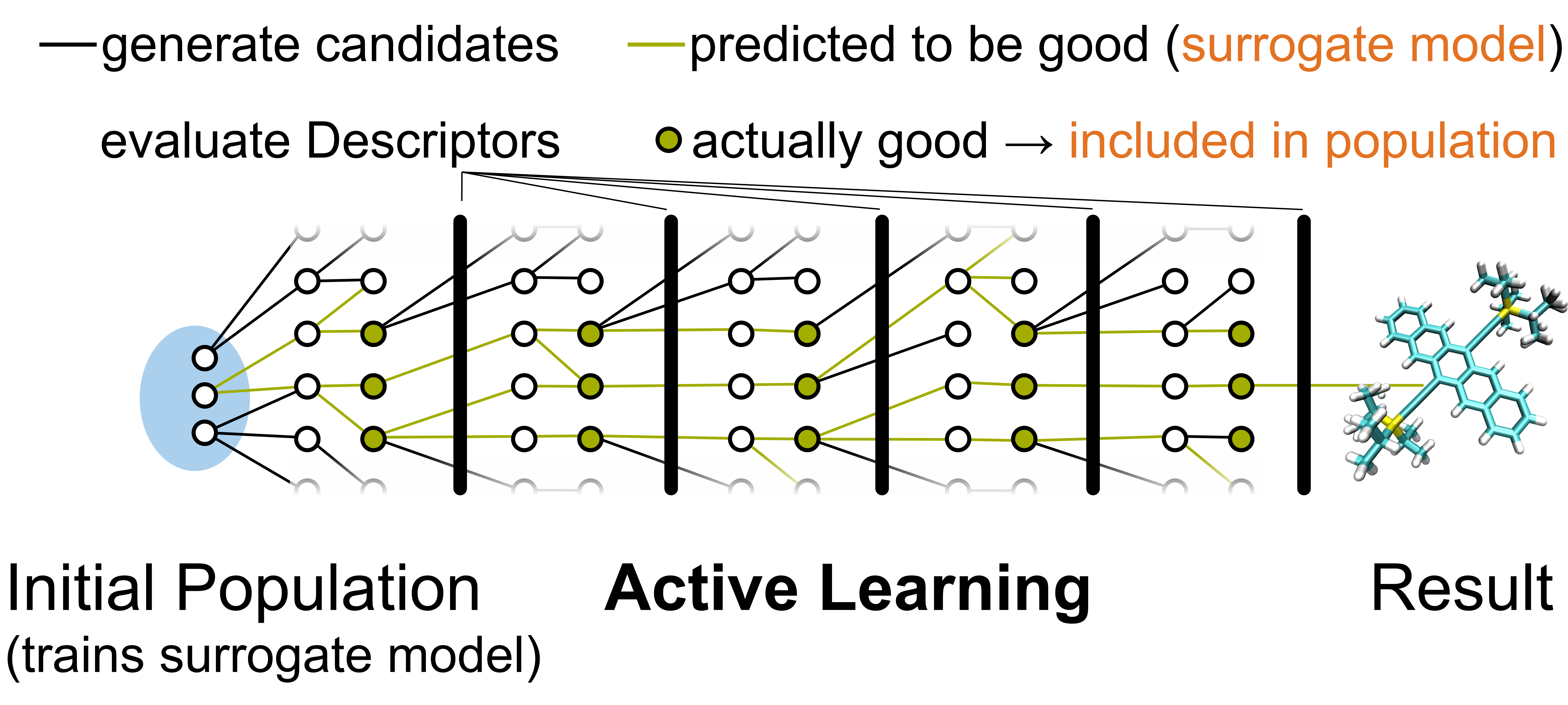
Considering for example organic electronics, the possible design space of small molecular crystals alone easily ranges in the tens of millions of compounds. This awards unparalleled flexibility of possible materials- or electronic properties where crystals can be tailored to fit any purpose. On the other hand, the huge variability is also the main factor hampering computational materials design, as the potential search spaces are enormous and not easily amenable to standard theoretical methods. We therefore work on the identification of good descriptors for e.g. the conductivity of organic solids and methods to determine them cheaply but accurately. With these we can then either in classical computational funnel schemes or employing surrogate models in an active machine learning approach not only identify good candidates for optimised materials but also generate vast amounts of data for further knowledge-based exploration of design space. Indeed, in our work we strive to extract general design criteria through a combination of advanced, relative visualisation techniques as well as mathematically sound statistical testing. Visualisation of chemical space networks thereby allows us to identify regions of chemical design space where promising examples have not yet undergone systematic improvements e.g. through synthesis of systematically modified molecules and crystals. In this regards, using our rigorously tested design rules, such as e.g. certain combinations of molecular building blocks, could be a way to fully explore design space regions so far under-utilized in experiments.
Charge Transfer
In recent years a new class of electronics based on organic molecules or polymers has appeared on the market. Most prominent examples here are of course organic light emitting devices (OLEDs), which found use in screen and lighting technologies, but also organic field effect transistors (OFETs) and even organic solar cells (OSCs) are found in more and more applications. The potential advantages of a technology mainly based on organic molecules are numerous, ranging from lower production costs compared to rare earth containing inorganic solids, to novel material properties allowing e.g. for flexible devices. Unfortunately, the mechanisms governing charge transport are not at all well defined, making them hard to tackle computationally. At the same time, interactions at interfaces e.g. between organic crystals and metallic electrode materials as well as charge transport across these interfaces are poorly understood. Furthermore, theoretical studies here tend to be hampered by the large system sizes and computationally intensive methods needed to accurately describe charge transport. A major part of our work is therefore devoted to the development of efficient, yet accurate methods to simulate the movement of charge carriers in organic semiconductors as well as across metal/molecule interfaces. Based on these methods we estimate so-called descriptors, computationally undemanding properties of system that are correlated with a quantity of interest, such as the charge carrier mobility. These we apply in large scale screening studies to generate databases materials properties for the extraction of structure function relationships and thus rules for the potential design of efficient organic semiconductors.
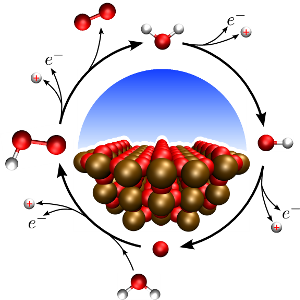
Photo-Electrocatalysis
Especially in an energy context, electro- and photo-electrocatalysis assume a more and more central. Large scale sustainable generation of energy carriers -- such as H2 and Methanol -- and at the same time reduction of CO2 are important targets in light of climate change and finite fossil resources. The feasibility of such an approach obviously rests mainly on the catalysts, their stability, availability and efficiency. In order to optimise existing catalysts and find completely new materials, an atomistic understanding of the specific reaction mechanics is of paramount importance and it is there that computational approaches can contribute to catalysis research. I our group, we study the pathways of common electrocatalytic reactions such as e.g. Water oxidation for H2 production and specifically test common assumptions and simplifications. To allow for an unbiased examination of pathways, that for example also includes defected catalyst surfaces and charged reaction intermediates, we develop embedding methods which remove finite size effects inherent to standard periodic-boundary condition simulations. Finally, we also investigate the influence of the solvent on reaction properties through a mixture of explicit and implicit solvation methods.
Dielectric Response Methods
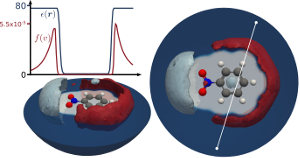
Efficient treatment of the dielectric response of a medium to perturbations, specifically the response of a solvent to a solvated molecule or cluster, is an important problem in modern theoretical chemistry. Explicit treatment of solvent degrees of freedom is very costly, as it necessitates a thorough statistical sampling for example through molecular dynamics (MD), and -- given e.g. the failures of density functional theory (DFT) in representing liquid water -- often not very accurate either. Another, more approximate approach rapidly gaining attention is therefore to replace explicit solvent molecules through a continuum model. This not only removes many degrees of freedom to keep track of but, depending on the application, possibly the need for MD simulations altogether. In this research focus, we develop and improve implicit solvation models for application with DFT. We generalise existing solvation schemes to account for interfaces and ionic effects to study their influence on chemical reactions such as photo-electrocatalysis or charge transport in organic electronics. As these are effective models we also investigate their validity in microscopic simulations.
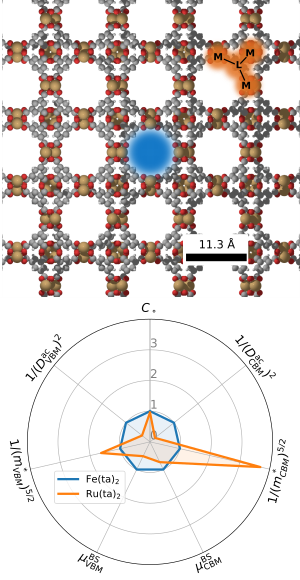
Metal Organic Frameworks
Metal-organic frameworks (MOFs) are a class of porous media, where organic linker molecules connect metal centres. Due to their well defined structure and comparatively cheap fabrication they have many potential applications ranging from gas-storage, to catalysis to applications in medicine and sensing. Due to the flexibility of the MOF concept with respect to the choice of organic linker molecules they can even be functionalised with, e.g. photo-switchable molecules. Depending on the exact functionalisation, certain MOF properties -- such as conductivity, elastic modules or pore size -- could be dynamically influenced through the application of light. These design freedoms make MOFs a hot topic in today's materials' research. Yet, especially concerning MOF-functionalisation, microscopic understanding still lags behind structural developments, with e.g. the photo-chemical reaction mechanisms or the aspects hampering charge transport being largely unknown. Here, theoretical studies offer a way to shed light on some of these questions. In our group, we employ a number of static and time-dependent ab-initio techniques to study possible reaction pathways of functionalised MOFs, as well as mechanisms of charge transport through certain MOFs. In the case of photo-switchable molecules, MOFs show a number of important differences to the gas or liquid-phase such as e.g. entropic barriers to some reaction pathways. On the other hand, only few MOFs have been found to experimentally exhibit non-vanishing conductivity, due to a variety factors hampering charge transport. These range from a low intrinsic carrier density, to large effective masses and electron-phonon coupling. All of these factors can, if estimated with sufficient computational efficiency, be used as descriptors in screening and design of more efficient MOFs.