Forschung
Batteriematerialien
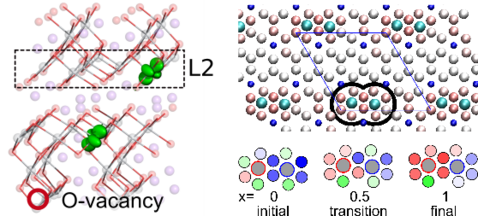
Angesichts des immer dringender werdenden Umstiegs auf ein CO2 neutrales Wirtschaftssystems ist neben den Problemen bei der nachhaltigen Energieerzeugung eine der größten Herausforderungen die effiziente Speicherung der so erzeugten Energie. Eine Option, elektrochemische Batteriesysteme, die im Prinzip seit über 200 Jahren bekannt sind, zeigen nur mangelhafte Energiedichten und Langzeitstabilitäten wie sie z.B. fossile Energieträger aufweisen. Um diese Limitationen zu überwinden werden auf der ganzen Welt innovative neue Konzepte wie z.B. Festkörperbatterien erforscht. Ein großer Teil dieser Forschungen bezieht sich dabei auf die Identifikation neuer Materialien und die Charakterisierung der beteiligten mikroskopischen Mechanismen um diese für maximale Effizienz einsetzen zu können. Aufgrund der Natur einer elektrischen Batterie, beruhen diese Mechanismen auf dem Transport von Ladungsträgern wie Elektronen oder Ionen. Über die Jahre hat unsere Gruppe eine Reihe von Methoden implementiert die sich perfekt dafür eignen die grundlegenden Prozesse die in Batteriematerialien auftreten zu untersuchen.
Diese mikroskopischen Einsichten können dann dazu dienen bereits bekannte Materialien weiter zu optimieren, z.B. durch Erzeugung von Defekten in spezifischen Mustern. Dadurch lassen sich Materialien erzeugen die nicht nur erhöhte Leitfähigkeiten sondern auch bessere Stabilitäten aufweisen.
Computergestütztes Materialdesign
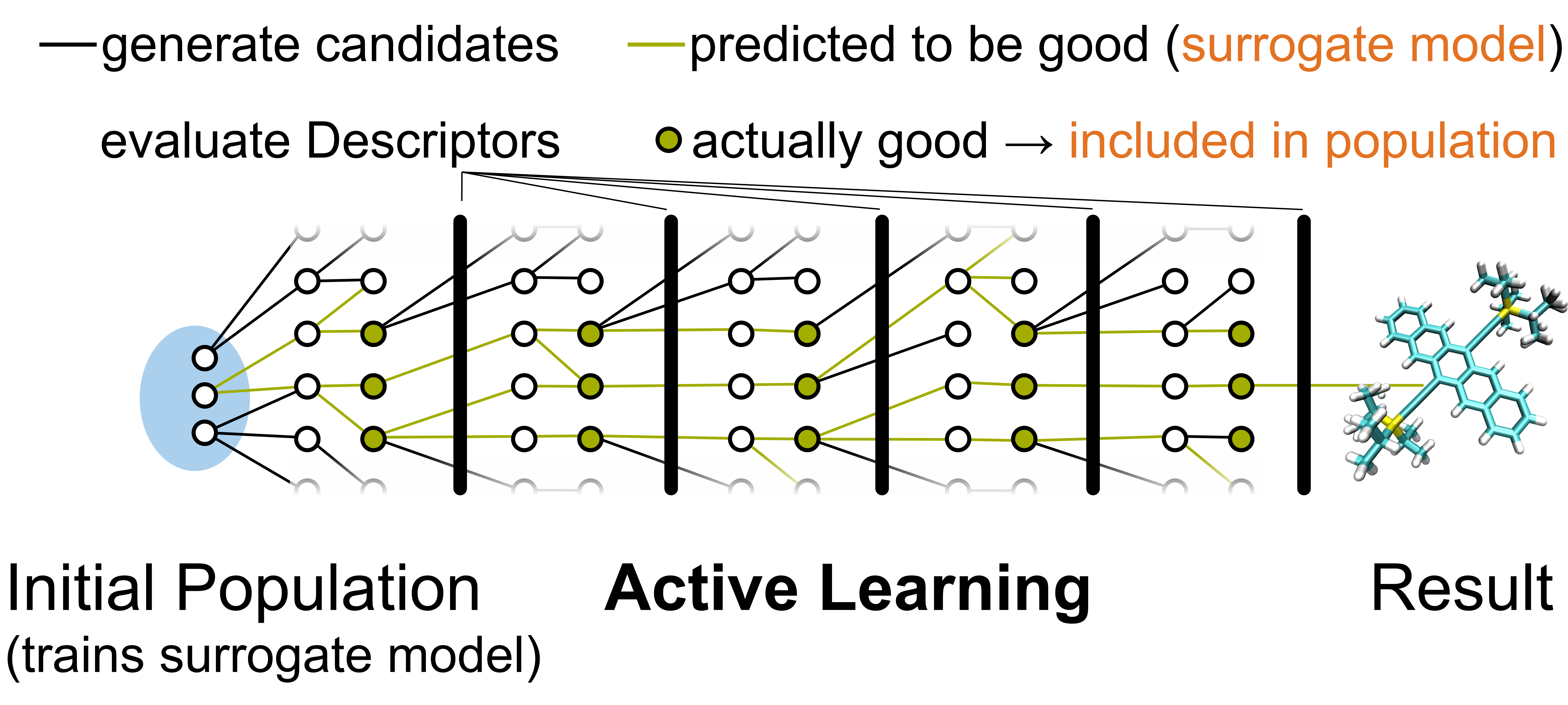
Ein großer Teil unserer Forschung ist das design neuer Materialien für verschiedenste Anwendungen. Jedoch ist, zum Beispiel für organische Elektronikanwendungen, der Designraum möglicher molekularer Kristalle riesig mit vielen Milliarden von möglichen Molekülen. Für die Anwendung entstehen dadurch unübertroffene Flexibilitäten bezüglich möglicher Material oder elektronischer Eigenschaften welche je nach Bedarf zurechtgezimmert werden können. Gleichzeitig, behindert diese große Variabilität die Identifikation neuer Materialien aufgrund der enormen Räume die theoretisch durchsucht werden müssten. Daher arbeiten wir an der bestimmung guter Deskriptoren, rechnerisch wenig fordernde Eigenschaften der Systeme, die mit den gewünschten Observablen, wie z.B. der Ladungsträgermobilität, korrelieren, sowie Methoden diese mit ausreichender Genauigkeit zu berechnen. Mit diesen können wir dann nicht nur vielversprechende Materialien in entweder klassischem in-silico screening oder mittels maschinengelernter Modelle identifizieren, sondern generieren auch enorme Datenmengen für die weitergehende Analyse des Designraums produzieren. In der Tat versuchen wir in unserer Arbeit generelle Designkriterien abzuleiten aus einer Kombination fortgeschrittener Visualisierungstechninken und mathematisch solider statistischer Tests. Visualisierungen erlauben uns dabei Zusammenhänge zwischen chemisch ähnlichen Materialien zu erkennen und so Bereiche des Designraums zu finden die bislang nicht ausreichend systematisch verbessert wurden, wie z.B. durch Synthese modifizierter Moleküle und Kristalle. Unsere rigoros getesteten Designregeln, wie z.B. bestimmte kombinationen von molekularen Bausteinen, können in dieser Hinsicht einen Weg zur vollständigen Erforschung des Designraums darstellen.
Ladungstransport
In den letzten Jahren sind nach und nach erste elektronische Geräte welche auf organischen Molekülen und Polymeren basieren am Markt erschienen. Prominente Beispiele dafür sind organische Leuchtdioden (OLEDs), zu finden in Bildschirmen und Beleuchtungsmitteln, aber auch organische Feldeffekttransistoren (OFETs) und Solarzellen (OSCs) finden mehr und mehr Anwendungen. Solche Technologien, basierend vornehmlich auf organischen Molekülen, haben zahlreiche Vorteile wie z.B. niedrigere Produktionskosten im Vergleich zu anorganischen Materialien die oft seltene Erdelemente beinhalten, oder neuartige Materialeigenschaften beispielsweise für flexible Geräte. Leider sind die Mechanismen welche Ladungstransport in diesen Materialien bestimmen nicht wohl definiert, was deren theoretische Behandlung verkompliziert. Zudem sind Wechselwirkungen an Grenzflächen, z.B. zwischen organischen Kristallen und Metallelektroden und Ladungstransport durch solche Grenzflächen bisher nicht gut untersucht. Dazu kommt, dass theoretische Studien solcher Materialien durch die notwendigen Systemgrößen und teuren Simulationsmethoden weiter erschwert werden. Daher ist die Entwicklung effizienter und dennoch genauer Methoden zur Beschreibung des Ladungstransports in organischen Halbleitern ein großer Teil unserer Arbeit auf diesem Gebiet. Diese Methoden erlauben es uns sogenannte Deskriptoren, rechnerisch wenig fordernde Eigenschaften der Systeme, die mit den gewünschten Observablen, wie z.B. der Ladungsträgermobilität, korrelieren. Diese Deskriptoren erlauben es uns wiederum, in sogenannten screening studien große Datenbanken von Materialeigenschaften zu erzeugen, welche es uns erlauben mithilfe statistischer Methoden Zusammenhänge zwischen Struktur und Funktion der Materialien zu extrahieren. Diese können dann dazu dienen rationale Designregeln for zukünftige organische Halbleitermaterialien abzuleiten.
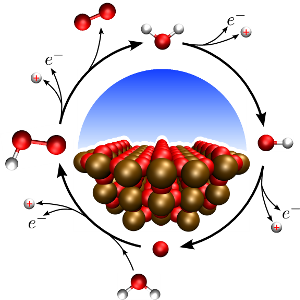
Photo-Electrocatalysis
Especially in an energy context, electro- and photo-electrocatalysis assume a more and more central. Large scale sustainable generation of energy carriers -- such as H2 and Methanol -- and at the same time reduction of CO2 are important targets in light of climate change and finite fossil resources. The feasibility of such an approach obviously rests mainly on the catalysts, their stability, availability and efficiency. In order to optimise existing catalysts and find completely new materials, an atomistic understanding of the specific reaction mechanics is of paramount importance and it is there that computational approaches can contribute to catalysis research. I our group, we study the pathways of common electrocatalytic reactions such as e.g. Water oxidation for H2 production and specifically test common assumptions and simplifications. To allow for an unbiased examination of pathways, that for example also includes defected catalyst surfaces and charged reaction intermediates, we develop embedding methods which remove finite size effects inherent to standard periodic-boundary condition simulations. Finally, we also investigate the influence of the solvent on reaction properties through a mixture of explicit and implicit solvation methods.
Dielektrische Response Methoden
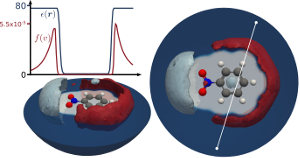
Die effiziente Behandlung der dielektrischen Antwort eines Mediums auf Störungen, spezifisch die Antwort eines solvents auf ein solvatisiertes Molekül oder Material, ist ein sehr wichtiges Problem in atomistischen Computersimulationen. Die Explizite Behandlung der Freiheitsgrade der Lösungsmittel ist rechnerisch sehr teuer, da damit ein eingehendes Sampling, z.B. mittels Moleküldynamik (MD) einhergeht und, wenn man z.B. die bekannten Fehler von elektronischer Dichtefunktionaltheorie (DFT) bei der Berechnung von flüssigem Wasser bedenkt, oft auch nicht sehr genau. Deshalb, findet ein anderer, approximativerer Zugang, bei dem explizite Lösungsmittelmoleküle durch Kontinuumsmodelle ersetzt werden, mehr und mehr Anwendung. Dadurch wird nicht nur die Zahl der Freiheitsgrade reduziert, sondern auch, je nach Anwendung, die Notwendigkeit von MD simulationen komplett eliminiert. Daher ist ein Teil unserer Forschung die Entwicklung und Verbesserung sogenannter Impliziter Lösungsmittelmodelle zur Anwendung mit DFT. Wir generalisieren bereits existierende Methoden um Übergänge zwischen dielektrischen Umgebungen und ionische Effekte zu simulieren und erforschen deren Einfluss auf verschiedenste reaktionen wie Photo-Elektrokatalyse oder Ladungstransport in organischen Halbleitern. Nachdem es sich bei diesen Methoden um Effektive Modelle handelt untersuchen wir abschließend auch deren Gültigkeit in mikroskopischen Simulationen.
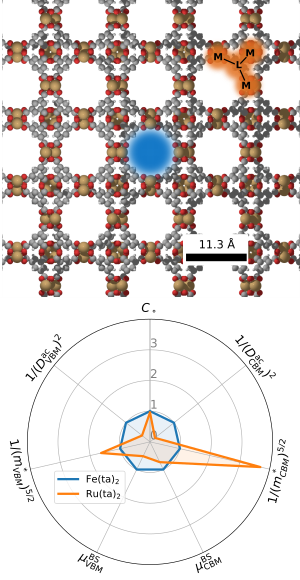
Metall Organische Netzwerkverbindungen
Metall Organische Netzwerkverbindungen (MOFs) sind oft poröse Festkörper welche aus organischen Verbindungsmolekülen bestehen die durch Metallzentren zusammengehalten werden. Aufgrund ihrer wohldefinierter Struktur sowie vergleichsweise einfache Herstellung haben MOFs viele potentielle Anwendungen, von Gasspeicherung über Katalyse zu Anwendungen in Medizin und als Sensoren. Aufgrund der Flexibilität des MOF Konzepts bezüglich Wahl der Linkermoleküle können MOFs sogar funktionalisiert werden, z.B mit photo-schaltbaren Molekülen. Abhängig von der exakten Funktionalisierung, können gewisse MOF Eigenschaften wie z.B die Leitfähigkeit, Elastizität oder Porengröße, sogar dynamisch durch die Applikation von Licht oder elektrischen Feldern beeinflusst werden. Genau diese große Designfreiheit macht MOFs zu einem interessanten Thema der modernen Materialforschung. Interessanterweise jedoch hinkt gerade auf dem Gebiet der MOF-Funktionalisierung das mikroskopische Verständnis den strukturellen Entwicklungen hinterher. So sind z.B. photo-chemische, piezo-elektrische, oder den Ladungstransport hindernde Faktoren großteils unbekannt. Gerade diese Fragen können jedoch durch theoretische Studien zumindest teilweise beantwortet werden. Unsere Gruppe bringt dazu eine Reihe von statischen und zeitabhängigen ab-initio Methoden zum Einsatz um mögliche Reaktionswege funktionalisierter MOFs sowie deren Ladungstransportmechanismen zu untersuchen. Im Fall der Funktionalisierung mit photo-schaltbaren Molekülen, gibt es z.B. wichtige Unterschiede zwischen Molekülen in der Gasphase und im MOF, beispielsweise durch entropische Barrieren durch das MOF Gerüst. Dazu kommt dass bislang nur wenige MOFs existieren die im Experiment nicht-verschwindende Leitfähigkeiten zeigen. Dazu tragen eine Reihe von Faktoren bei wie z.B. niedrige Ladungsträgerdichten, hohe effektive Ladungsträgermassen und starke Elektron-Phonon Kopplung. Jeder dieser Faktoren kann daher als Deskriptor für das Screening und Design von effizienteren MOFs verwendet werden.